
Why New Medical Device Regulations Require More Than Ad-Hoc Manual Processes

This time around, products really do have to ‘do what they say on the tin’!
Reducing Errors In Factory Print: The Case For Labeling Digitalization

It is too risky to leave the accuracy of market-facing product information to chance
UDI & Product Traceability For Medical Devices: White Paper

How a more strategic approach to labeling digitalization can better serve businesses & regulators
Lessons & Leftover Challenges from MDR Preparations: What Next?

How medical device companies can effectively overcome the challenges posed by EU MDR
EU MDR Labeling Compliance - Are We There Yet?

Bob Tilling, VP Global Sales, explains how medical device manufacturers can recover lost time
How Companies Are Using New EU MDR Regulations To Improve Artwork & Labeling Processes

EU MDR forced changes on medical device manufacturers but has led to improvements in multiple areas
Key EU MDR Changes You Need To Know About

New medical device regulations substantially change the ways manufacturers market devices in the EU
What We Can Learn From EU Medical Device Regulation Compliance Preparations

MDR requirements alone should not determine how manufacturers overhaul their label management
Is Your Medical Device Organization On Track To Comply With EU MDR?

The clock is ticking on the need to ensure compliance with the EU Medical Device Regulation (EU MDR)
EU MDR: An Opportunity For Medical Device Manufacturers

Compliance is not a short-term business overhead, it is an opportunity to improve best practice
Labeling and the Art of Data Quality for UDI
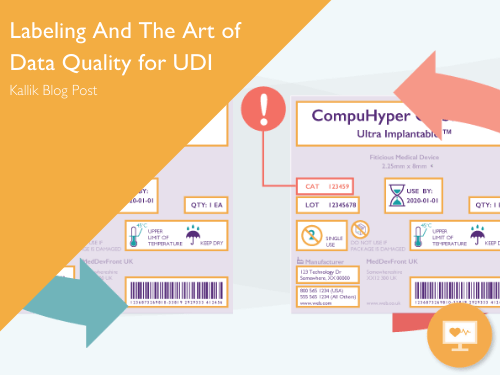
How adopting best practice for managing labeling content can bring business value to UDI projects
Why UDI Is Europe's Latest Hot Topic

Not only does data have value, but structured data has increased value
How To Improve Return On Investment From UDI

The transition to UDI is a huge undertaking, so it is important companies take a wider perspective
Factory Printing: How To Reduce Errors, Accelerate Printing Times and Maintain Compliance

The risks of using manual processes for factory printing and how to successfully tackle them
Factory Labeling Migration Following Business Acquisition

Veraciti, with 21 CFR Part 11 electronic signatures, eliminated paper-based quality processes
Veraciti™ Sets New Quality Benchmarks at Integra LifeSciences

Surgical and medical instrument manufacturer Integra LifeSciences is a leader in medical technology
3 MDR Lessons Class II Should Learn From Class III Medical Device Businesses

Why it is vital medical device manufacturers begin planning a technology-led, long-term solution?
6 Steps Medical Device Manufacturers Must Take When Navigating The Digital Maturity Curve

For medical device manufacturers faced with tight regulations, manual processes are not feasible
Helping Device Manufacturers Get Those Vital Ventilators Built

Delivering ventilators to the NHS on time with required labeling during the peak of the pandemic
How To Ensure Full IVDR Compliance On Time

A guide on how your company can get compliant with new medical device regulations
4 Key Actions To Ensure IVDR Compliance Deadlines Are Met

The next challenge after EU MDR is IVDR. Don't let your business fall short of compliance
Why Digital Maturity Is Essential For Medical Device Manufacturers

Digital maturity is paramount in highly regulated industries, and we’re going to explain why
Why Your Business Should Act Now To Be Compliant With IVDR

Why your business should act now to be compliant with IVDR
Kallik Brings Technology & Industry Expertise To Help Teleflex Deploy A Global Labeling System

How Kallik helped Teleflex revolutionize their labeling
How Managing The Simple Medical Device Label Could Make Or Break Nordic Expansion Ambitions For Man

How effectively managing the simple device label could be key to overcoming the challenges of entering the
Leading Orthopaedics Company Össur Overcomes IFU Management Challenges With Veraciti™ Solution

Overhauls legacy asset management systems and disjointed processes to improve global business operations an
